


The Jackson Laboratory Center for Precision Genetics Advances Therapeutics for Rare Diseases
Enabled by ongoing advancements in genome editing technology, precision disease modeling is an increasingly important component of personalized medicine approaches to treat human disease. In 2014, ORIP at NIH launched the Precision Disease Modeling Initiative (PAR-14-280) and funded three U54 Pilot Disease Modeling Centers in 2015. The initial cohort’s success (see Archives Information on the Initial Cohort) led ORIP to renew this initiative in 2020 (PAR-20-085) and award three new U54 Centers. The Jackson Laboratory (JAX) Center for Precision Genetics (JCPG) is one of the three centers established to provide the biomedical community with the advanced animal models needed to develop precision therapies for monogenic and complex disorders. ORIP modified its Precision Disease Modeling program to emphasize the formation of pipelines to support nominations from the research community to generate disease-specific human genomic variants for cost-effective, high-throughput screening in animal models.


Dr. Cathleen (Cat) Lutz, Vice President, Rare Disease Translational Center (RDTC), JAX, (Figure 1) and Dr. Stephen (Steve) A. Murray, Professor and Senior Director, Genetic Resource Science, JAX, (Figure 2) co-lead JCPG (U54OD030187).
JCPG’s aim is to create mouse models and build a pipeline for phenotyping and drug testing with a focus on models that carry the exact genetic mutations responsible for certain rare diseases. Drs. Lutz and Murray agree that their research should directly benefit patients. That is why—although programs that focus on validating variants of unknown significance within the rare disease population are important—JCPG prioritizes the increasing number of individuals with confirmed genetic diagnoses. Given recent advances in gene therapies, it is easy to imagine how this research can be parlayed into meaningful results for patients.
Now in its fifth year, JCPG has generated nearly 70 mouse models, with more in progress, and has supported more than 30 families and foundations. Dr. Murray remarked that “NIH/ORIP funding has provided the resources to create that critical element, which is the model that everybody wants and needs.” While industry is tackling rare diseases like Duchenne muscular dystrophy and cystic fibrosis—both of which have larger patient populations and are more likely to provide a financial return on investment—JCPG uniquely addresses ultra-rare diseases. Fewer patients have each disease but they collectively represent a very large unmet need. Drs. Lutz and Murray have responded to that need by concentrating on modeling known pathogenic mutations in mouse models. JCPG collaborated with The University of Texas Southwestern Medical Center researchers to generate a mouse model for enoyl-CoA hydratase, short chain 1 deficiency (ECHS1D), a rare pediatric disease with no effective treatments that results in severe developmental delays, encephalopathy, hypotonia, and early death. The ECHS1D mouse model revealed that altered metabolic signaling and inflammation may contribute to epilepsy, which was attributed to impaired valine metabolism.1 Dr. Lutz explained that “understanding the mutation and mechanism of diseases allows us to design both mouse models and therapeutic strategies aimed toward individualized medicine.”
Many family and patient organizations request the services of the JAX RDTC, an overarching program that includes the JCPG. Typically, patient organizations, clinicians, or researchers contact the RDTC with a specific model need, which the Center then reviews through a nomination process. The JCPG proposes a strategy that evaluates the feasibility and timeline of the project. At each step in the process, the accepted projects and their organizers receive progress updates. Dr. Murray remarked that mouse models are the foundation of JCPG’s work and that the Center is looking ahead to potential targeted, gene-based therapies. The goal is to provide a preclinical future for each project that will move potential therapies closer to the clinic and ultimately help patients. This approach takes advantage of JAX’s extensive expertise in genetic engineering—inserting human DNA sequences in addition to the mutation—which can include entire gene replacements. “Humanizing” the model improves its translatability for genetic therapies that could be applied to rare and ultra rare diseases, including antisense oligonucleotides (ASOs) and genome editing.
One such disease is alternating hemiplegia of childhood (AHC), which is caused by rare genetic mutations that are thought to be more prevalent in the population than is currently recognized. As diagnostic sequencing has expanded, more patients with AHC are being identified, revealing a clearer picture of disease manifestation across this patient population. In AHC, mutations in the ATP1A3 gene result in developmental delays, intellectual disabilities, and seizures and dystonia events that threaten the patient’s health. To generate functional models that recapitulate the human disease, JCPG genetically engineered specific patient mutations along with the surrounding human DNA sequence, inserted these sequences into a mouse2, and developed a system of assays to induce dystonia events like those seen in patients. Next, they worked with collaborators on various therapeutic strategies, including ASOs. This humanized, translatable AHC mouse model is expected to inform a U.S. Food and Drug Administration pre-investigational new drug (IND) application to advance this research to the clinic.


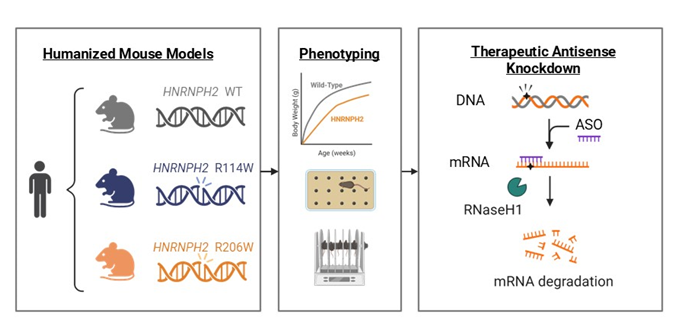
At the heart of every JCPG study are the patients and families who directly participate in and benefit from the work (Figure 3). For instance, JCPG is working with Casey McPherson, the founder of To Cure a Rose Foundation and the parent of a child who suffers from heterogeneous nuclear ribonucleoprotein H2 HNRNPH2 (Figure 4). HNRNPH2 is an X-linked disorder characterized by developmental delays, autism spectrum disorder, intellectual disability, motor and language delays, and seizures. Approximately 100 people have been diagnosed with an HNRNPH2 mutation, but some estimates show that as many as 10,000 people could be living with this condition.
Most mutations occur in a de novo manner with a dominant gain-of-function mechanism. To test potential therapies for this disease, JCPG created mouse models with patient-specific mutations in the HNRNPH2 genes, R206W and R114W (Figure 5) that carry human DNA sequences surrounding the pathogenic mutation. The engineered mice develop key phenotypes that reflect patient clinical presentation, including deficits in cognition and poor motor coordination. Such models allow therapeutics tested in mice to proceed swiftly to human clinical trials. The ASO knockdown therapeutic strategy is based on a compensatory paralogous gene, HNRNPH1, that increases in expression when HNRNPH2 is suppressed.
Although JCPG’s primary focus is building models for conditions with clear diagnoses, the Center also supports nominations for functional validation of novel variants. JCPG has reevaluated its process for engineering mouse models to be able to rapidly respond to the needs of human genetics investigators who use highly efficient CRISPR-Cas9 genome editing to directly characterize edited “F0” founder animals, which eliminates the need to breed mice. Because this method has focused on corroborating the causality of novel variants associated with structural birth defects, JCPG directly examines edited embryos, which greatly reduces the time required to generate functional data.
One successful application of this method was done in collaboration with Dr. Joseph Gleeson at the University of California, San Diego. The team modeled two variants of TMEM161B, a mutation that has been identified in a human population and was thought to be responsible for polymicrogyria (PMG), a disease characterized by abnormal folding of the brain and abnormal cortical development, but functional evidence to establish the connection has been lacking. Using the F0 method, the team corroborated the role of patient-specific mutations in PMG in a few short months,3 a timeline that would not be possible using traditional mouse modeling approaches. The team is now evaluating whether this approach can be applied to additional disease areas.
Drs. Lutz and Murray take great pride in this and other significant collaborations. Many families have already identified key research collaborators when they submit a nomination to JCPG, and Drs. Lutz and Murray make it clear that they have no intention of taking over any researcher’s academic projects but are collaborators, facilitators, and inventors who lead these projects at JAX. They advocate open access to data, resources, and information sharing via the ORIP-funded Mutant Mouse Resource & Research Centers (MMRRC) to advance science and research. JCPG also requires that its animal models be shared in the public domain through the MMRRC, in part because crowdsourcing information can make resources available more quickly to patients and their parents. Publications are the preferred method of dissemination, but they take time to produce. Dr. Lutz explained, “We want to treat the patients in front of us today, not just the next generation of children who are affected.” Accomplishing this requires mobilizing and moving quickly to engage collaborators and at the same time, train the next generation of researchers. Drs. Lutz and Murray have drawn on the knowledge of existing NIH programs, including the NIH Common Fund’s Knockout Mouse Phenotyping Program (KOMP2). With the skills and expertise for handling the different mouse models being developed and those gained from KOMP2 and from the Center’s characterization pipeline, they have carefully controlled phenotyping assays that are repeatable and cross-comparable.
JAX’s RDTC has grown thanks to JCPG’s successes. Dr. Lutz noted that the funding supplied by this grant is critical to the program’s success. However, the demand for downstream therapeutic testing can put a strain on precious dollars, so she and Dr. Murray worked to secure additional funding sources. JCPG applied for and received additional funding to support its work, including from the Chan Zuckerberg Initiative and the NIH Common Fund’s Somatic Cell Genome Editing program. In addition, each JCPG project has the potential to become the foundation for a new NIH R01 proposal. The current challenge is to keep the active projects, which patient foundations and their collaborators rely upon, moving forward. According to Dr. Lutz, it has been “really remarkable to see … how we have evolved from the initial concept that was more of a vision … to a reality that helps rare disease patients and their families.”
Going forward, JCPG will continue to review patient nominations, generate and validate mouse models, test therapies, file pre-IND applications, and raise funds for toxicology and manufacturing components. Mouse models generated by this program are publicly available and can be used for more therapeutic development and testing, including ASOs, gene editors, and, eventually, therapeutic strategies that target a common variant in an allele and allow researchers to treat multiple patients regardless of their individual mutations. “The beauty of the JCPG program is the investment in the mice that will exist in perpetuity, always there for the next generation of therapies to be tested,” added Dr. Lutz.
The Precision Disease Modeling program is one of ORIP’s major initiatives. For more information, please visit ORIP’s Precision Disease Modeling website.
References
1Eller MM, Zuberi AR, Fu X, et al. Valine and inflammation drive epilepsy in a mouse model of ECHS1 deficiency. Preprint. bioRxiv. 2024;2024.06.13.598697. doi:10.1101/2024.06.13.598697.
2Terrey M, Krivoshein G, Adamson SI, et al. Alternating hemiplegia of childhood associated mutations in Atp1a3 reveal diverse neurological alterations in mice. Preprint. bioRxiv. 2024;2024.12.24.630241. doi:10.1101/2024.12.24.630241.
3Wang L, Heffner C, Vong KI, et al. TMEM161B modulates radial glial scaffolding in neocortical development. Proc Natl Acad Sci USA. 2023;120(4):e2209983120. doi:10.1073/pnas.2209983120.